Projects
Enhancing Biomedical Question-Answering with a Keyword Frequency dirven Retrieval-Augmented Approach
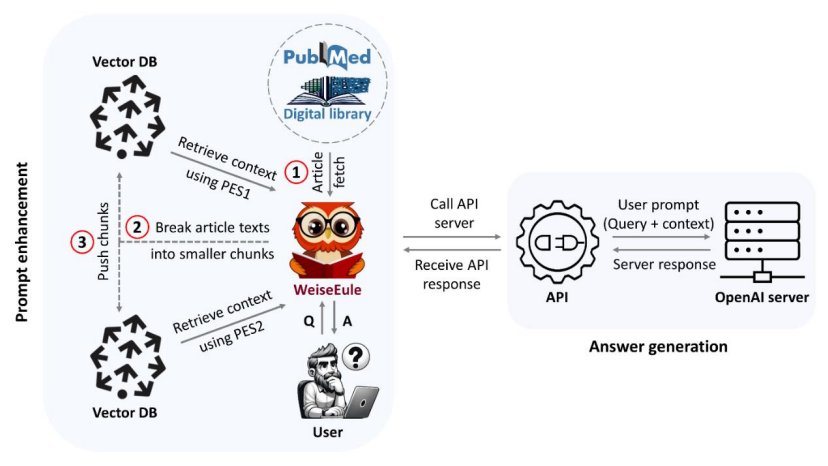
In this project, we tackled the challenge of extracting accurate and relevant information from the vast and complex biomedical literature, a task complicated by the field's specialized jargon and rapid evolution. While transformer models have revolutionized natural language processing (NLP), especially in large language models (LLMs) like GPT-4, they still struggle with generating up-to-date and accurate responses in specialized domains such as biomedicine. Our solution focuses on WeiseEule, an advanced QA bot we developed that leverages a retrieval-augmented architecture to guide LLMs in producing meaningful and accurate responses. We introduced a novel method that enhances prompts using explicit signals from user queries to extract the most relevant context, outperforming traditional text embedding-based approaches. We rigorously tested our method on 50 challenging biomedical questions, demonstrating that it significantly improves retrieval performance, achieving a Precision@10 of 0.95—indicating that the vast majority of the top results were highly relevant. Additionally, when evaluated for answer quality, our method outperformed the baseline and other models with a median quality score of 2.5 out of 3. WeiseEule also offers advanced features for review writing and identifying relevant articles for citation, making it a powerful tool for researchers. This work showcases the potential of prompt enhancement techniques to improve the reliability and accuracy of LLM-generated responses in specialized fields like biomedicine. By addressing key issues such as "hallucination" and outdated responses, WeiseEule provides a robust solution for biomedical QA, empowering users with greater control over the information generated.
Proteomic Data Analysis Pipeline based on LIMMA
This project involves the development of a user-friendly, open-source pipeline in R for conducting powerful and stable statistical analysis of proteomic data, particularly for two-group comparisons. The pipeline leverages the LIMMA (Linear Models for Microarray Data) method, an empirical Bayes approach, to enhance the detection of significant changes in protein abundance. By shrinking a protein's sample variance towards a pooled estimate, LIMMA provides more reliable inferences compared to traditional t-tests. The pipeline is designed to be accessible, automatically installing all required packages when executed in RStudio. It offers two modes of analysis—either including all proteins or excluding those with zero intensities in one group, allowing for flexible data interpretation. The exclusion mode helps focus on proteins with meaningful expression across groups, while still retaining the excluded proteins for additional analyses like Gene Ontology or pathway enrichment, enriching the biological context of the findings. The pipeline also includes robust normalization options to ensure data consistency, and it outputs results as interactive volcano plots and comprehensive TSV files. These plots, generated using both LIMMA statistics and ordinary t-tests, provide clear visualizations of the results, aiding in the identification of differentially expressed proteins. This tool is a valuable asset for researchers in proteomics, offering a streamlined and efficient way to conduct sophisticated statistical analyses and gain deeper insights into protein expression data. Compatible with major operating systems, it ensures broad accessibility and ease of use in various research environments.

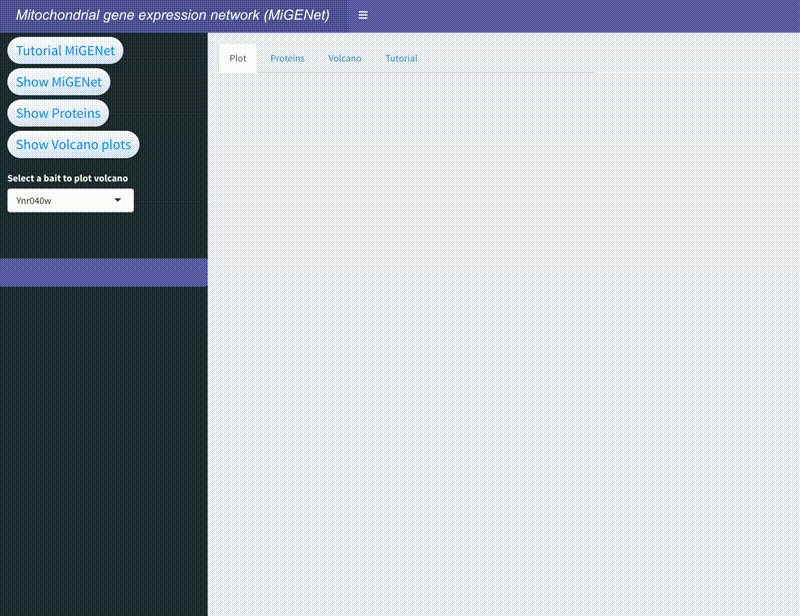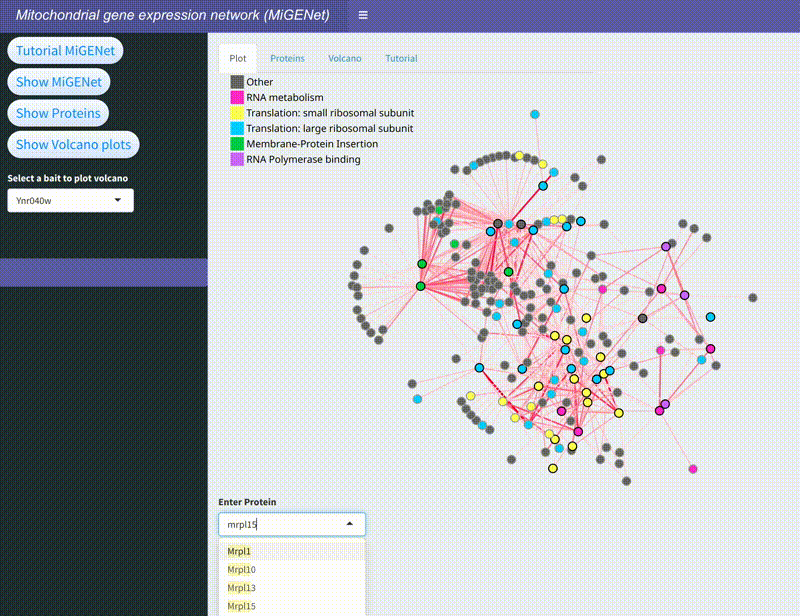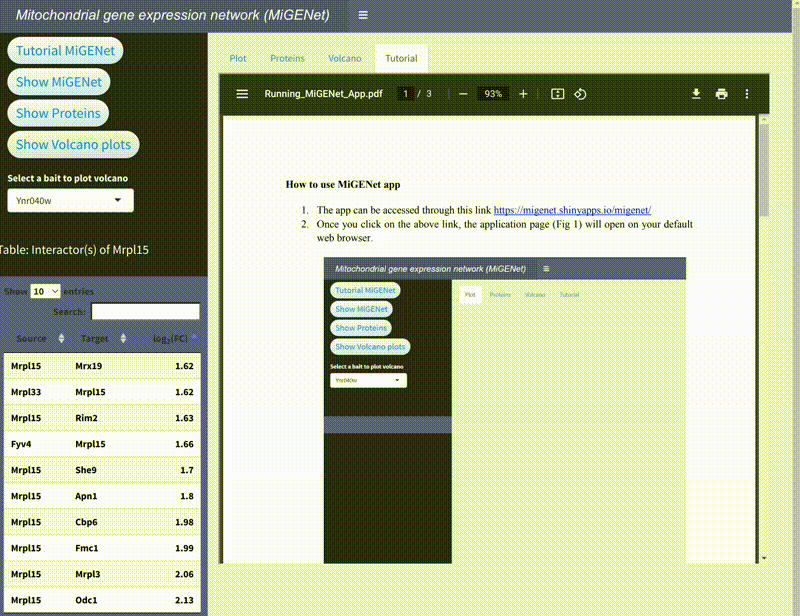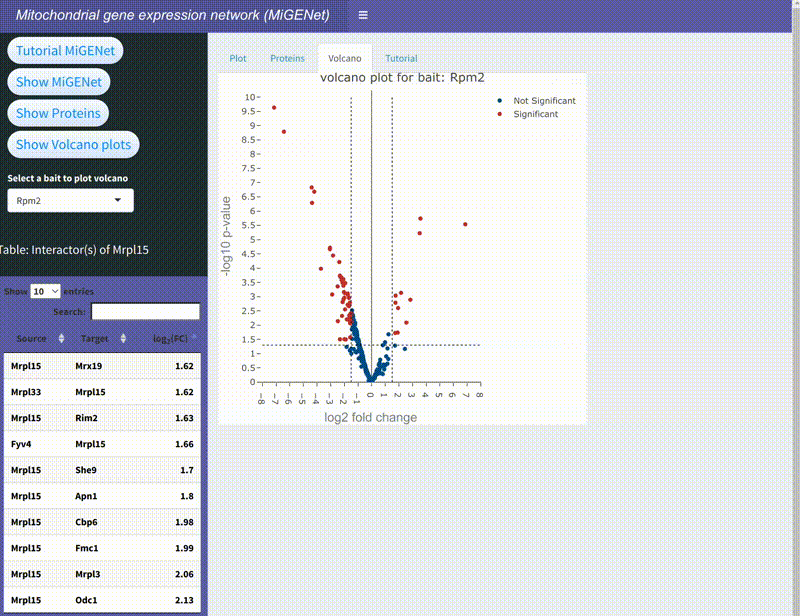
MiGENet: Mapping the Mitochondrial Gene Expression Network
In this project, we explored the intricate gene expression systems within mitochondria, focusing on the synthesis of key components of the oxidative phosphorylation (OXPHOS) complexes. Using the proximity-dependent biotinylation technique, BioID, combined with mass spectrometry, we identified and mapped a comprehensive network of proteins involved in mitochondrial gene expression in baker's yeast. This network, which we named MiGENet, includes factors responsible for transcription, RNA processing, translation, and protein biogenesis. Our analysis revealed the spatial organization of these processes, uncovering fundamental mechanistic principles and identifying proteins at critical sites within the mitochondria. Notably, we discovered that newly synthesized proteins are efficiently transferred from ribosomes to factors at the ribosomal tunnel exit, which are responsible for membrane insertion, co-factor acquisition, and integration into OXPHOS complexes. This coordination occurs at a specialized early assembly hub, ensuring the proper assembly and function of the mitochondrial OXPHOS system. The MiGENet project provides valuable insights into the unique and highly specialized nature of mitochondrial gene expression, offering a detailed understanding of how these vital processes are interconnected. This work advances our knowledge of cellular bioenergetics and has potential implications for understanding mitochondrial-related diseases.
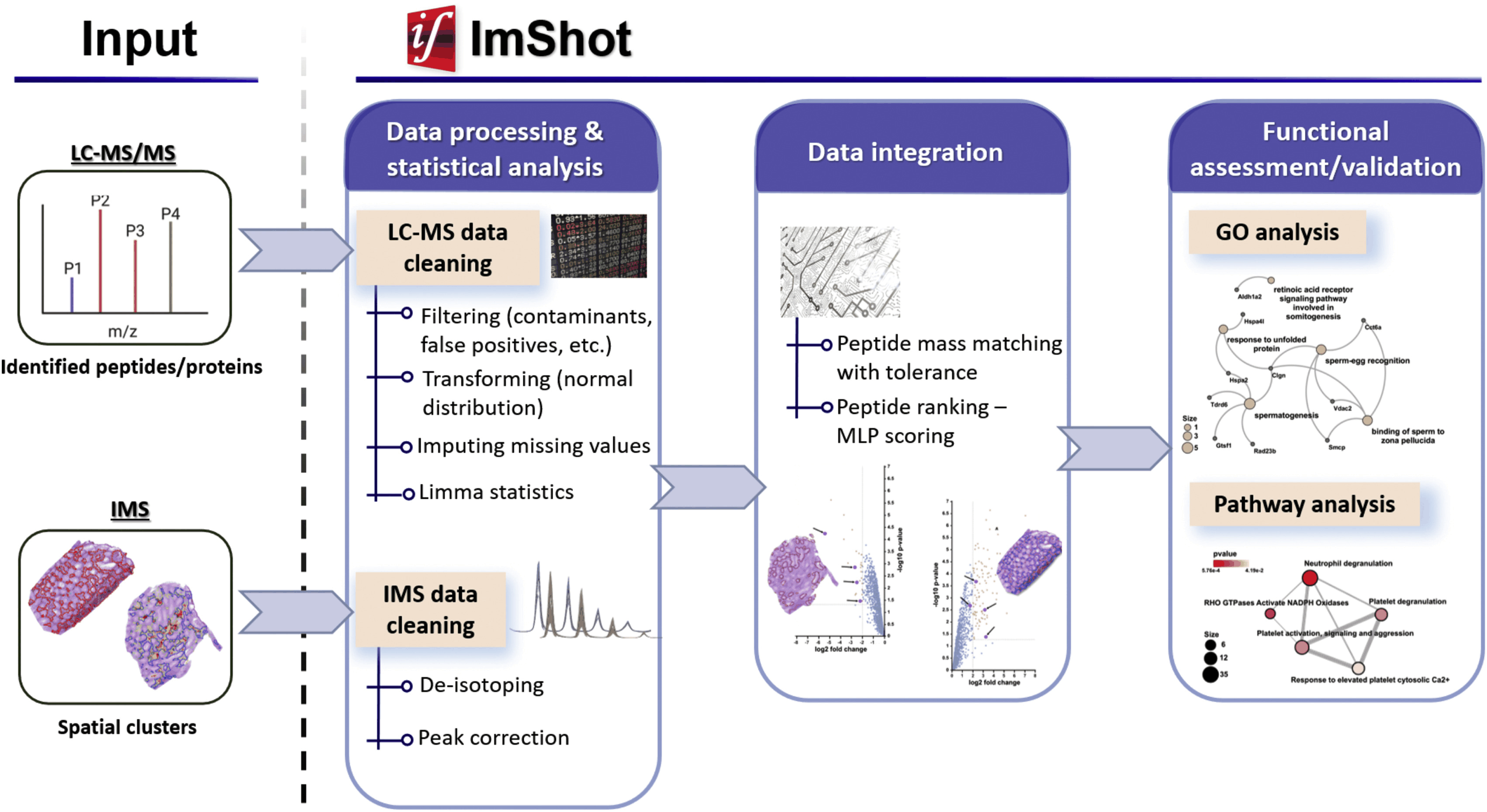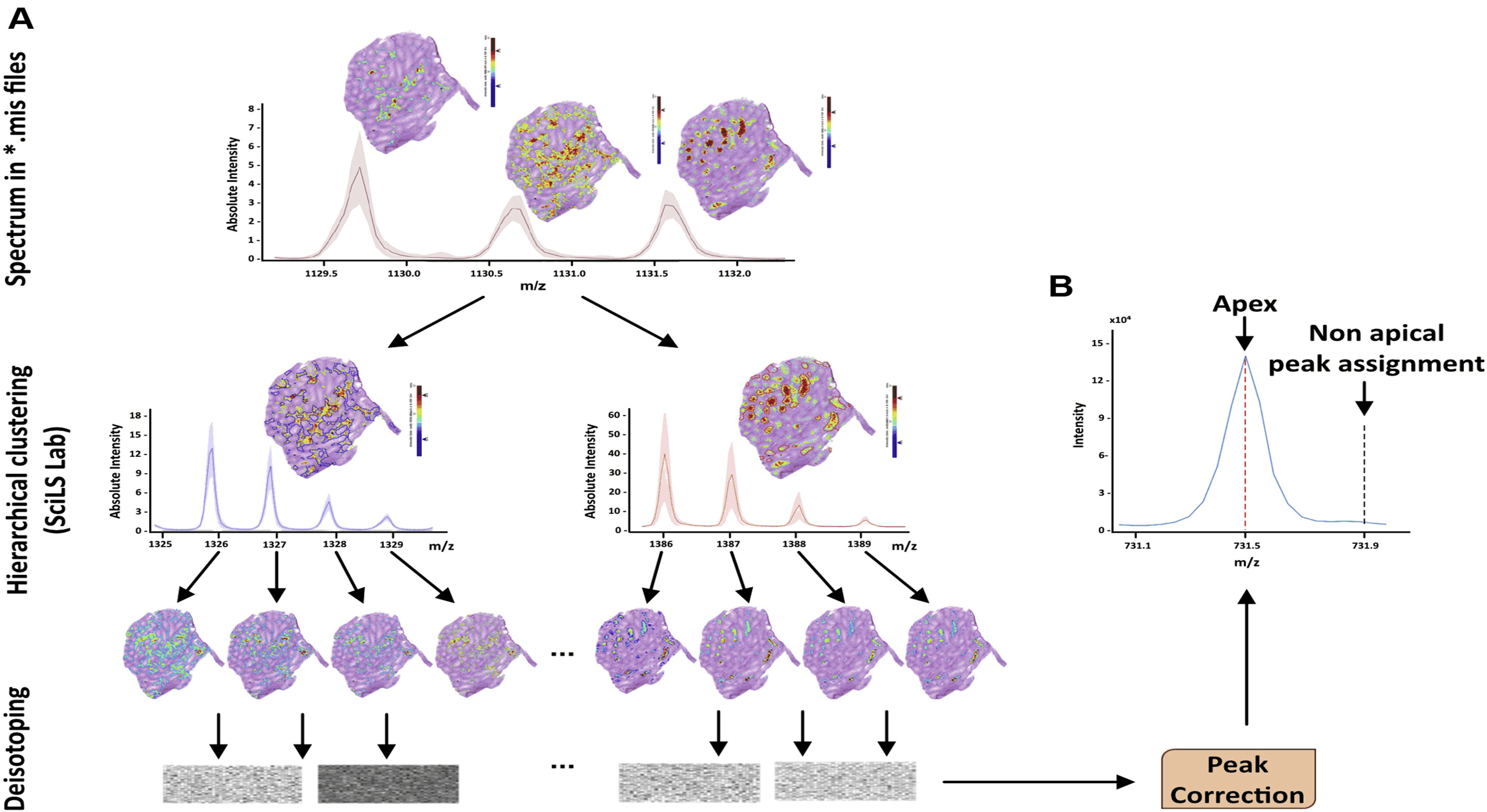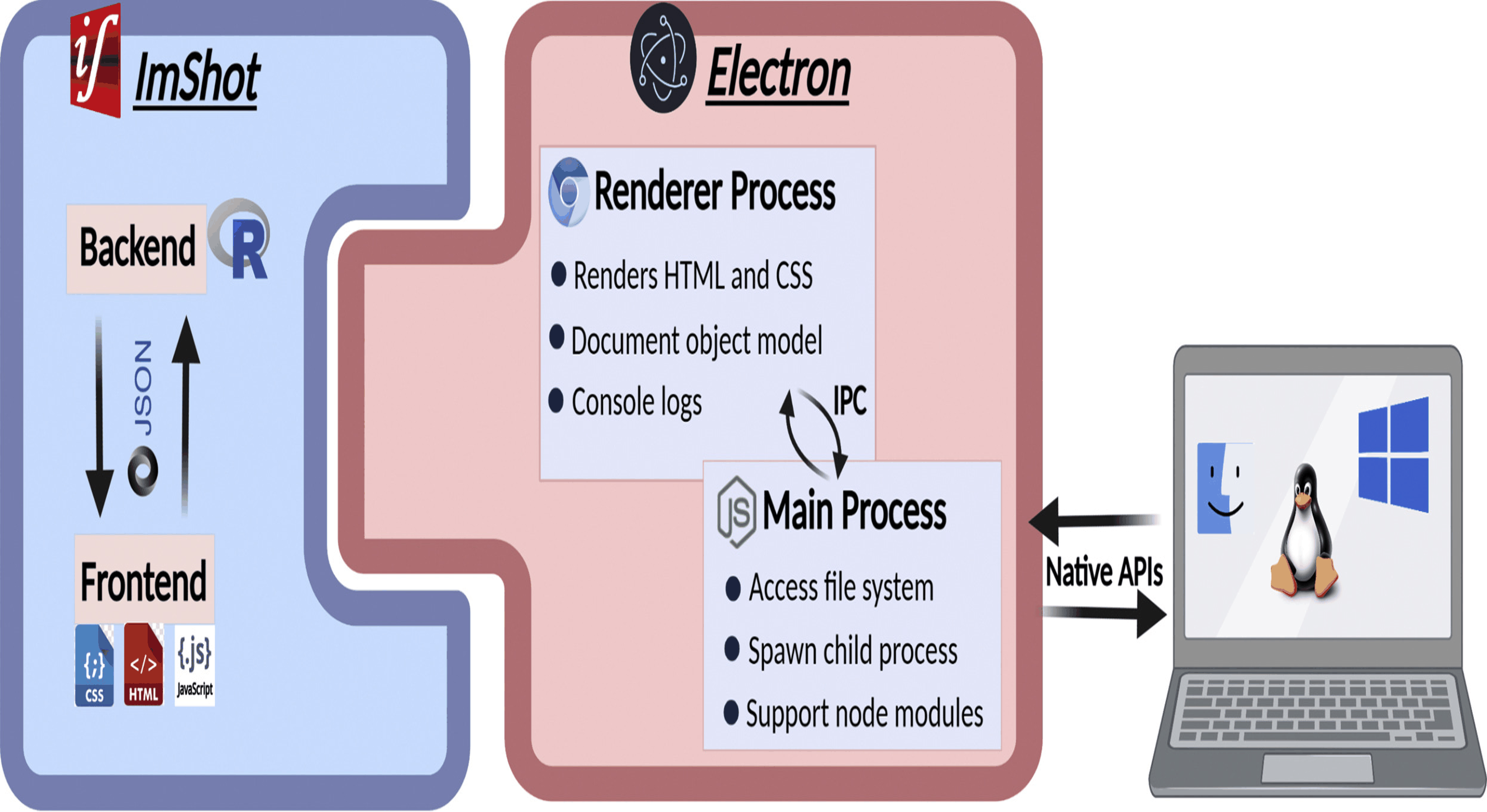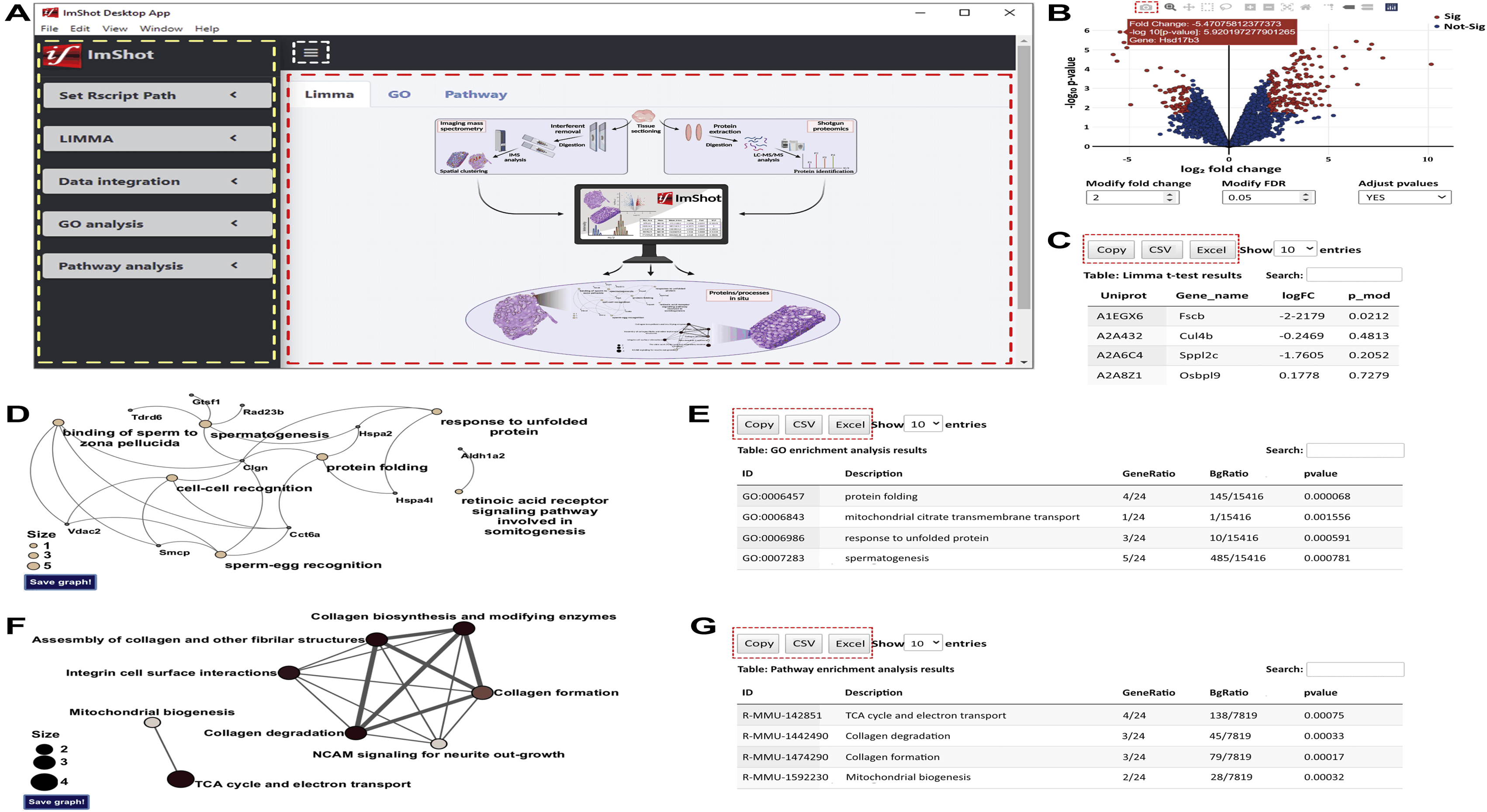
ImShot: Advancing Peptide Identification in Imaging Mass Spectrometry
This project led to the development of ImShot, a groundbreaking open-source software designed to tackle the challenges of peptide identification in MALDI Imaging Mass Spectrometry (IMS). Unlike traditional proteomics, IMS allows direct detection of proteins and peptides from biological tissues, providing valuable clinical insights. However, identifying these molecules has been a significant hurdle, often requiring complex and resource-intensive experimental setups with limited success. ImShot addresses this gap by integrating data from IMS with shotgun proteomics (LC-MS) of serial tissue sections. It employs a novel scoring system to accurately match detected peptides with their parent proteins, significantly improving identification accuracy. The software's modular design not only facilitates seamless data analysis but also enables users to handle LC-MS data independently, offering interactive visualizations like enrichment plots and Gene Ontology term analysis. Built with a user-friendly interface, ImShot is compatible with all major desktop operating systems and is freely available under the MIT license, making it an accessible and powerful tool for the scientific community. This software represents a significant advancement in the field of mass spectrometry, enabling more precise molecular identification and accelerating research in clinical diagnostics.
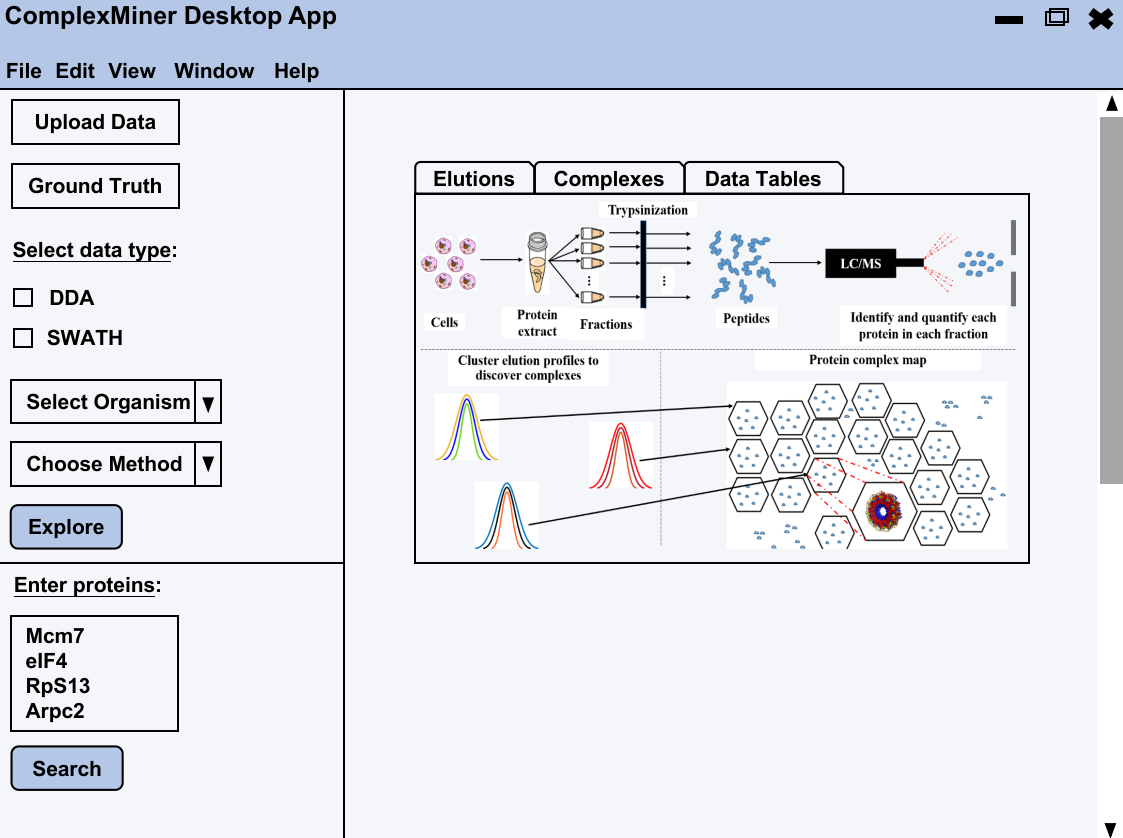
ComplexMiner: A User-Friendly Platform for Protein Complex Discovery
Understanding protein interactions is crucial for deciphering cellular functions, as protein complexes mediate nearly all biological processes within a cell. During my thesis, I developed ComplexMiner, a desktop application designed to simplify and enhance the discovery of protein complexes. Traditional methods for predicting these complexes typically rely on extensive machine learning models that require large, labeled datasets from multiple experimental modalities, leading to significant time and resource demands. ComplexMiner addresses these challenges by employing one-shot learning, a cutting-edge deep learning approach that allows for the identification of protein complexes with fewer experimental runs. This innovation not only reduces the experimental burden but also accelerates the discovery process. Recognizing the needs of the proteomics community, where most researchers are wet lab scientists, I designed ComplexMiner with a graphical user interface (GUI) to provide an intuitive and aesthetically pleasing platform. Unlike command-line tools, this GUI-based application allows users to explore complex datasets, perform sanity checks, and generate dynamic visualizations of extracted information with ease. Although ComplexMiner is still in the development and testing phase, my goal is to release it as an open-source tool, making it widely accessible to researchers and contributing to the advancement of proteomics research.
Enhancing Radial Basis Function Networks with Cosine Kernels
In this project, we introduced a novel approach to Radial Basis Function (RBF) artificial neural networks (ANNs) that significantly improves their ability to approximate functions and classify patterns. Traditional RBFs rely on Gaussian kernels based on Euclidean distance (ED) to measure the similarity between feature vectors and neuron centers. Our innovative method combines this with a cosine kernel that considers the angle between vectors, offering a new perspective on distinguishing features. We demonstrated that this combination can effectively distinguish between feature vectors in scenarios where conventional methods fall short. A practical application of our method is in adaptive symbol detection for multiple phase shift keying (MPSK) signals, where angle information is crucial. By testing our enhanced RBF across different domains, we found that it consistently outperforms traditional RBFs, offering a substantial improvement in accuracy and performance. This work not only introduces a novel computational technique but also has practical implications for signal processing and pattern recognition.
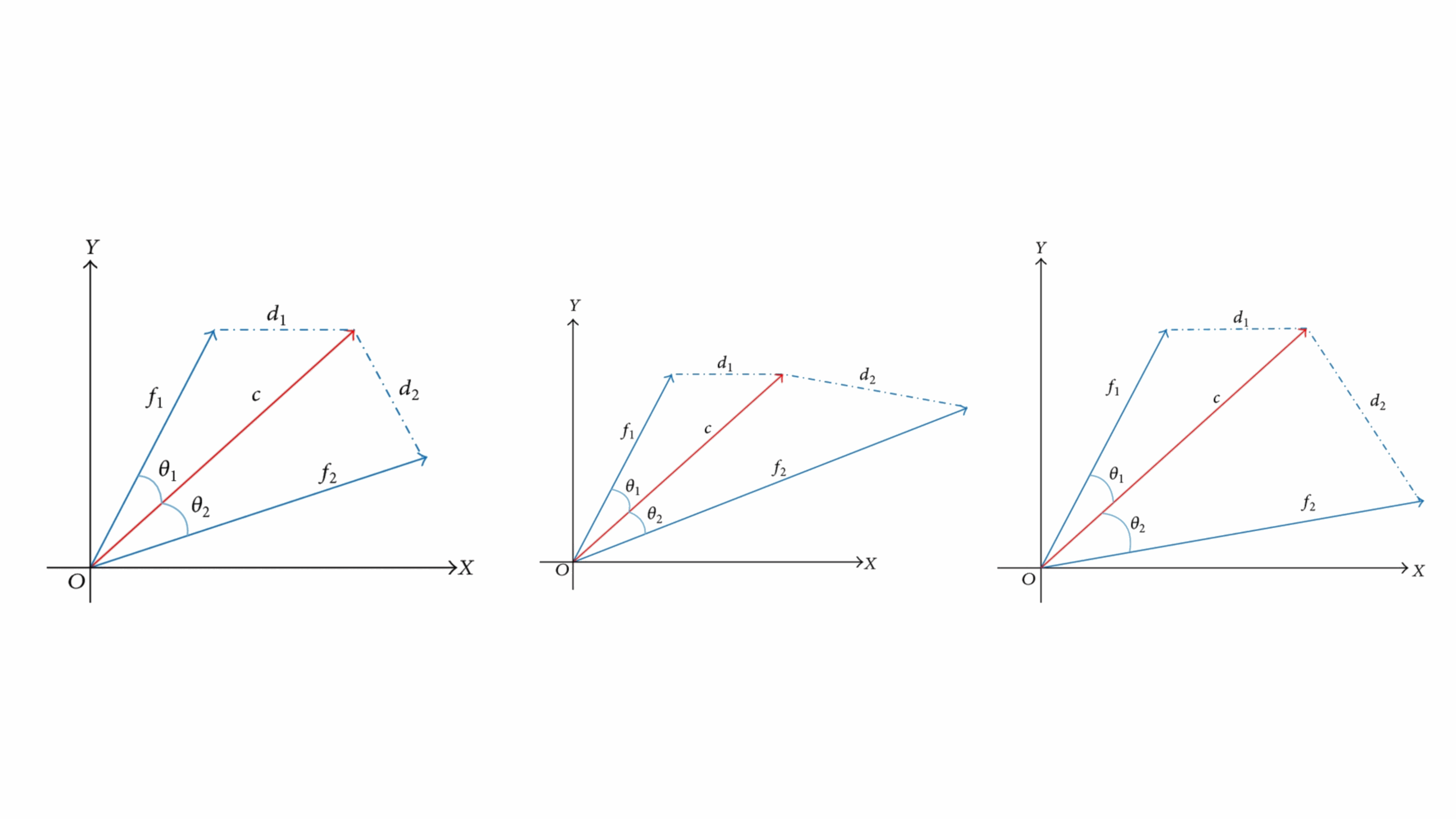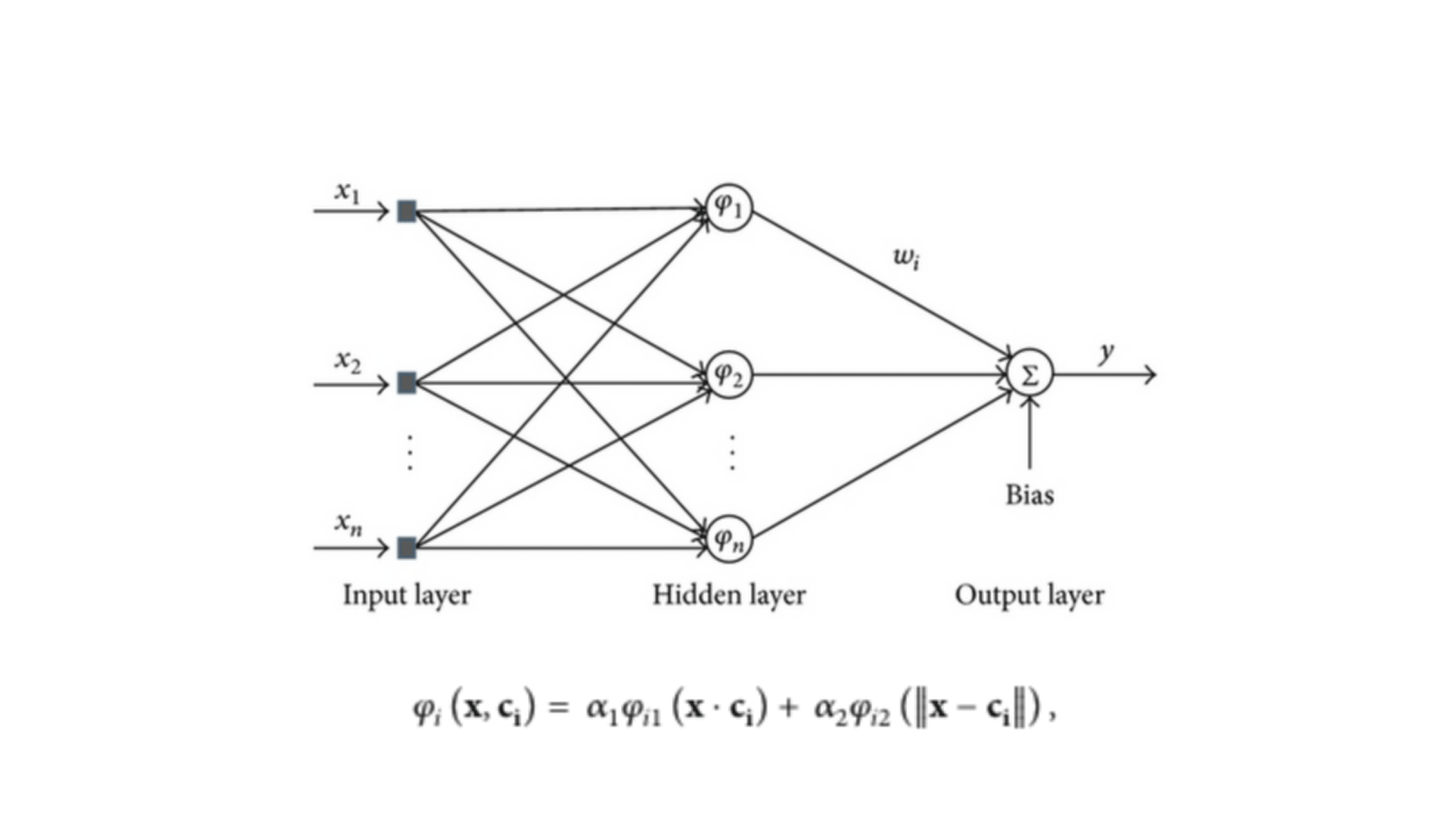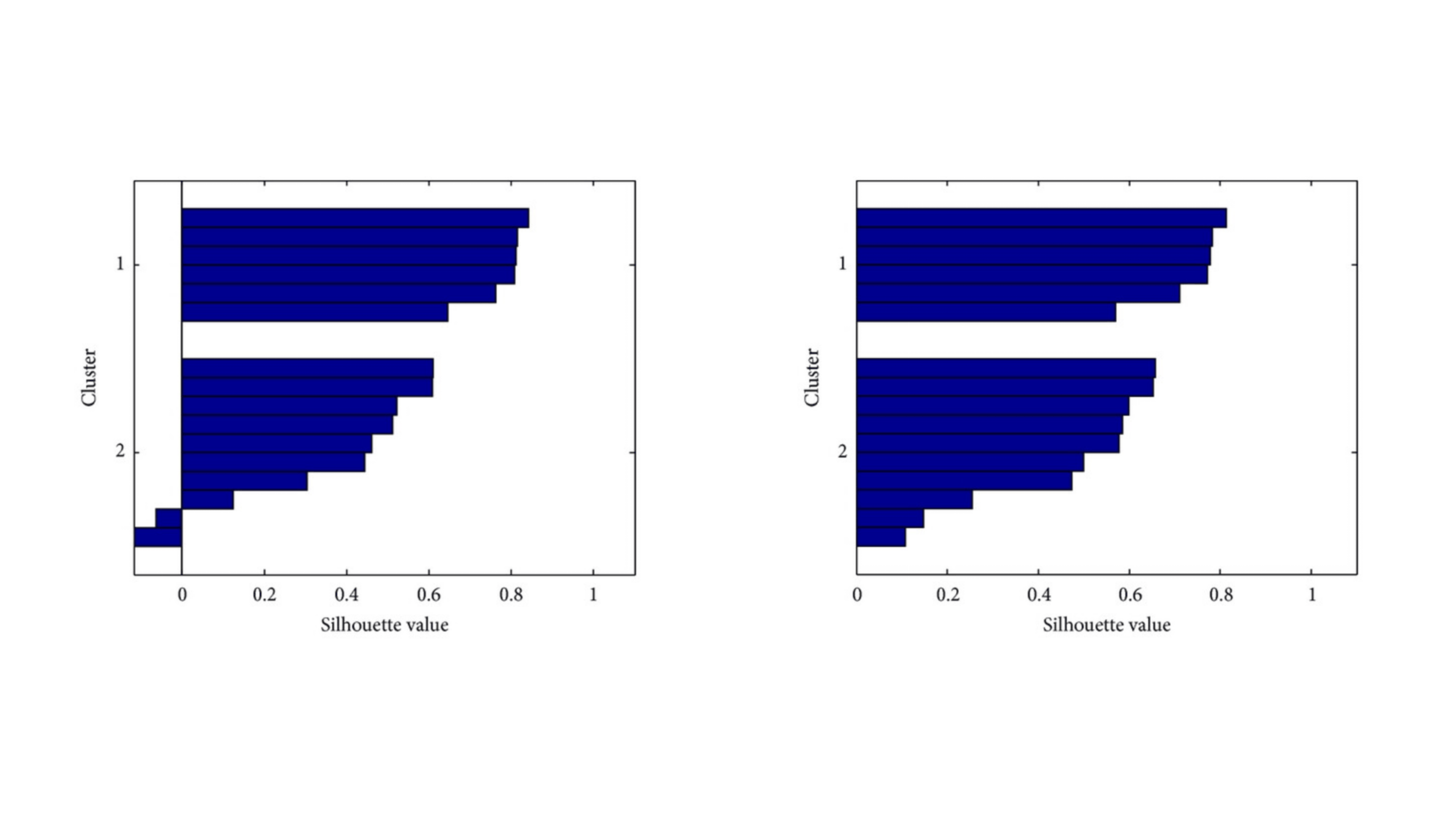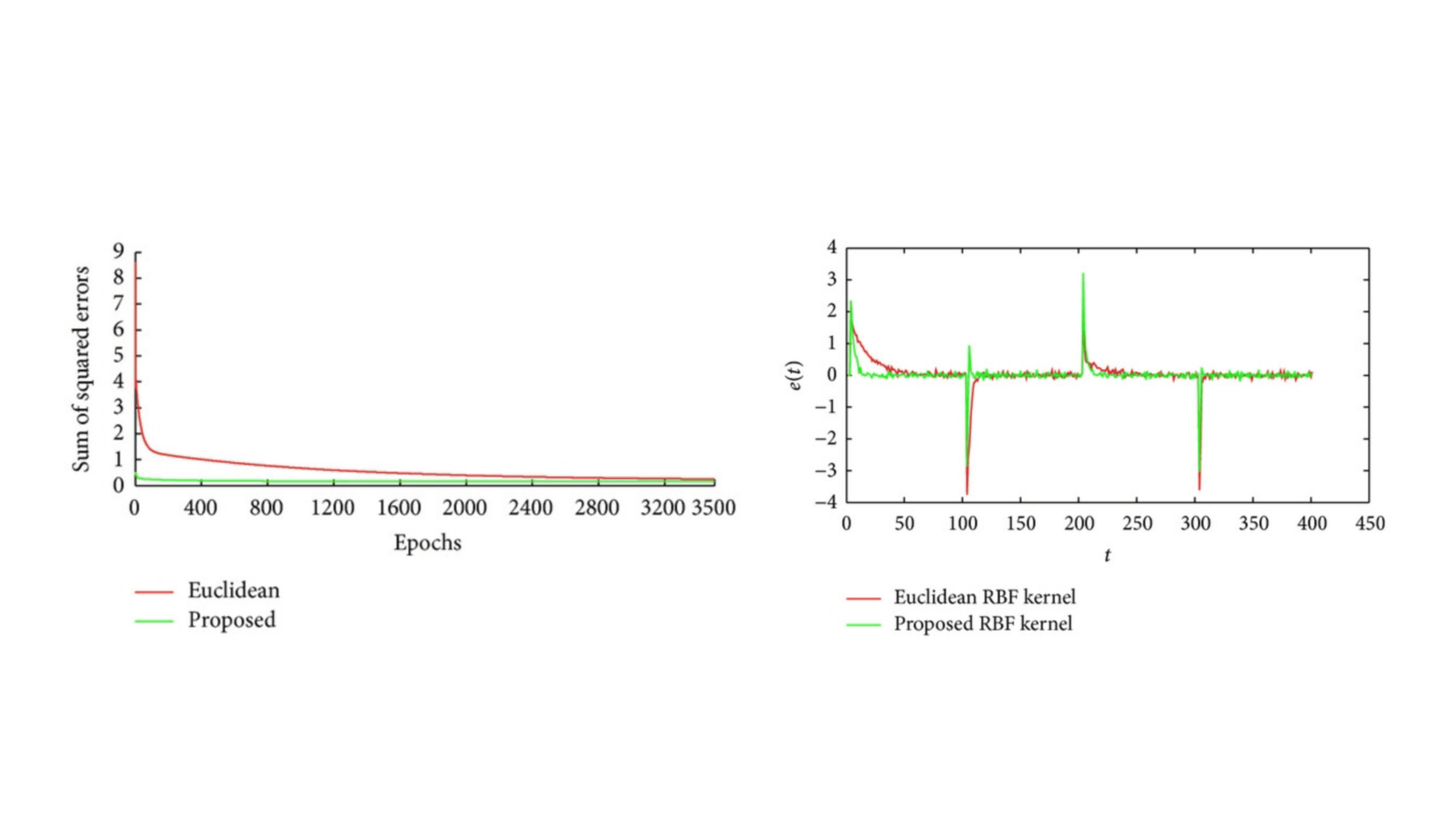
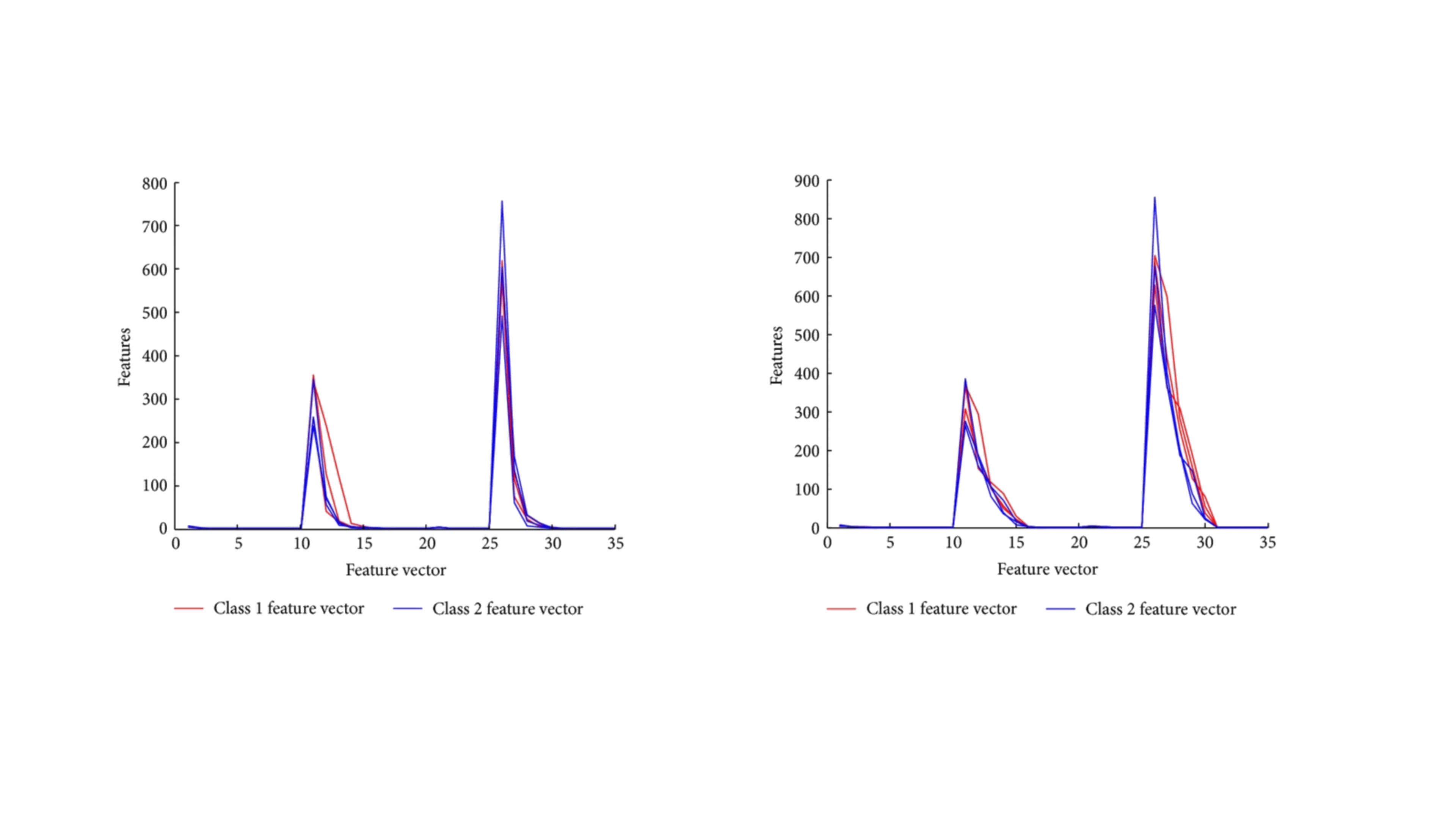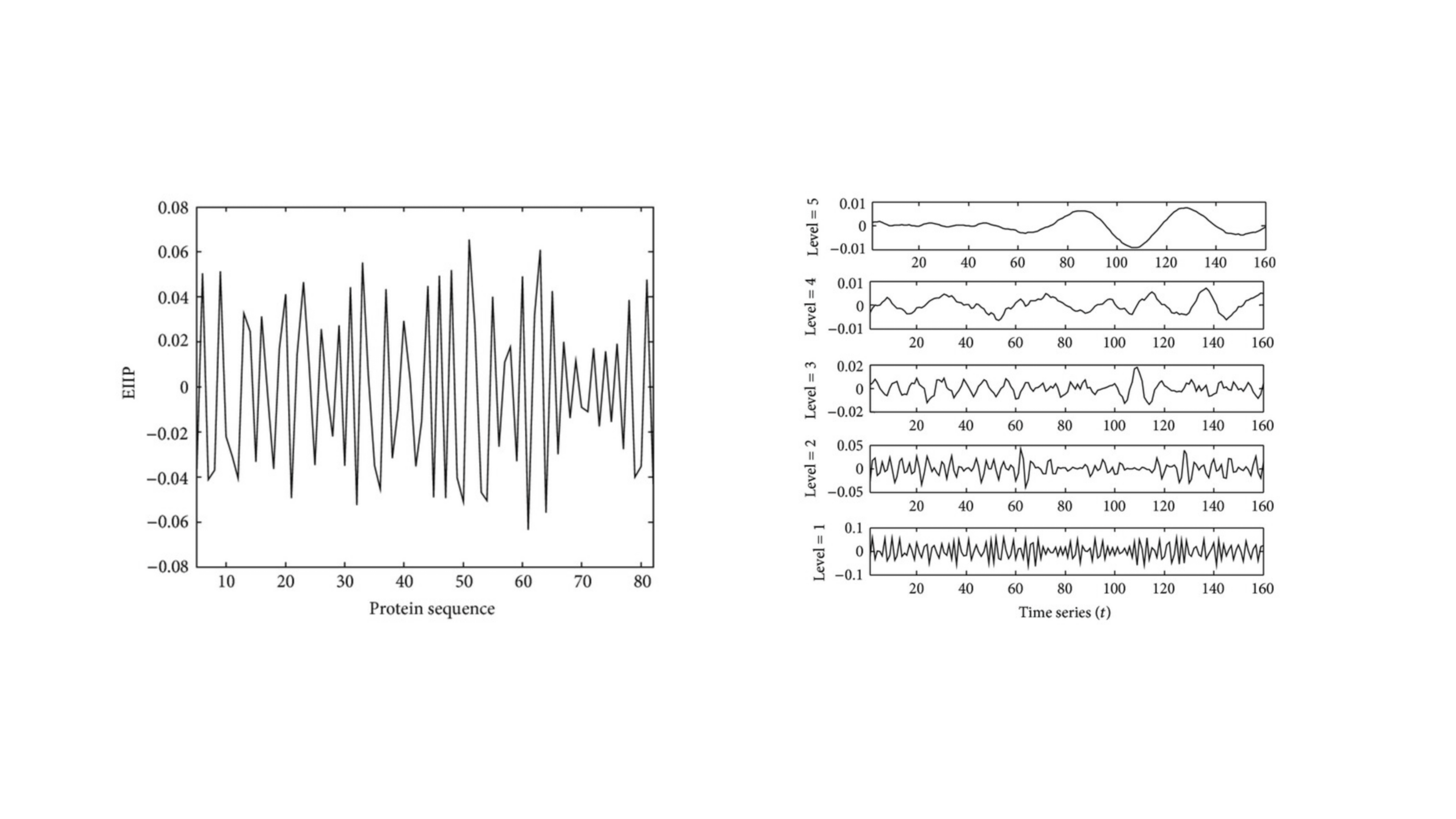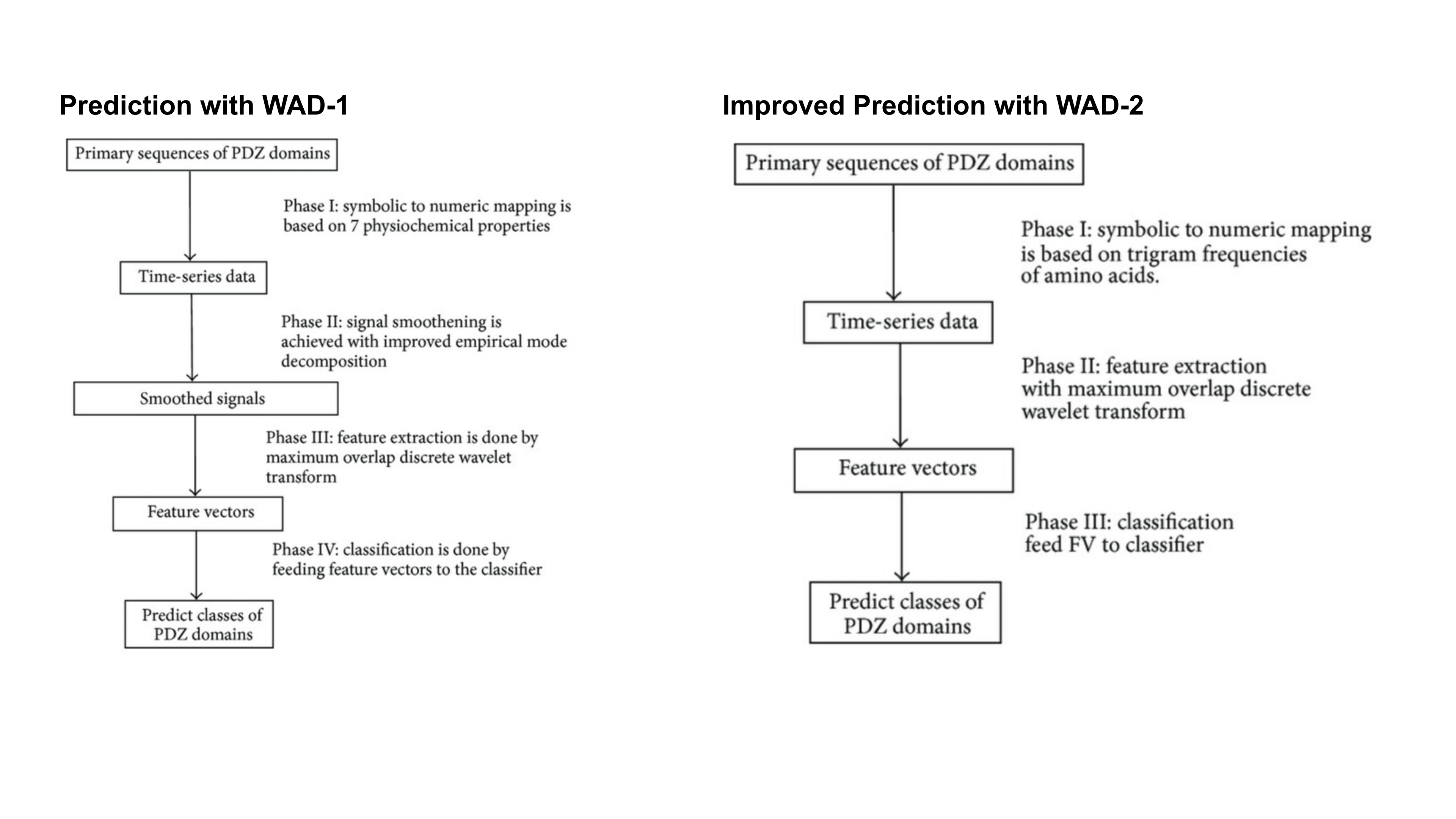
Wavelet-Based Classification of PDZ Domains
In this project, we developed innovative wavelet-based methods to classify and differentiate between Class I and Class II PDZ domains—key protein regions implicated in various diseases. PDZ domains are common in protein structures and understanding their classification is crucial for biomedical research. We introduced two computational methods, with one method surpassing standard wavelet techniques in recognition accuracy. Our models not only demonstrated impressive results but also hold potential as powerful tools for efficiently screening and reducing the search space in protein analysis. This advancement could significantly enhance the accuracy of identifying relevant protein structures, offering a valuable approach in the field of computational biology.